Alterações do material genético
Albinismo
Albinismo é a ausência parcial ou total do pigmento melanina na pele, no cabelo e nos olhos.
Pessoas de pele clara e rosada, olhos azul acinzentados e cabelos esbranquiçados sofrem de albinismo, uma patologia congénita em que os pais, não necessariamente albinos, são portadores do gene causador da doença.
Existe mais de um tipo de albinismo e podendo só atingir os olhos. No entanto, a forma mais perigosa de albinismo é a que determina a total ausência de pigmento por todo o corpo denominada albinismo universal.
O albinismo universal caracteriza-se por uma acentuada hipopigmentação da pele, cabelos e olhos. A pele é branco-leitosa e desenvolve eritemas intensos em resposta à exposição ao sol. O cabelo é branco, amarelado ou pardo-amarelado. Os olhos não têm pigmento na coróide nem na retina, e a íris é diáfana, em geral azul-acinzentada. Invarialvemente ocorrem nistagmo, fotofobia e diminuição da acuidade visual. As pupilas são às vezes vermelhas crianças, mas nos adultos são sempre negras.
De uma forma geral, não há meios de prevenir o albinismo.
Casais de famílias com histórico da doença devem passar por um trabalho de aconselhamento genético par evitar que seus filhos nasçam albinos. Não são necessários testes para diagnosticar o albinismo, pois os sintomas são bem visíveis. A doença de forma alguma modifica ou diminui a expectativa de vida de seus portadores, mas o quotidiano de quem tem albinismo é limitado por causa da intolerância à luz solar.
O cancro de pele e a cegueira são as maiores ameaças para essas pessoas, o que exige um monitoramento constante desses riscos.
Anemia Falciforme
As hemoglobinopatias constituem um grupo de doenças genéticas decorrentes de anormalidades na estrutura ou na produção da hemoglobina, molécula presente nos glóbulos vermelhos que leva oxigénio a todas as partes do corpo.
As doenças hereditárias causadas por alterações na hemoglobina afectam milhões de pessoas em todo o mundo. Entre elas destaca-se a anemia falciforme, resultante de uma mutação no gene que codifica a cadeia globina b, levando à formação da hemoglobina de falsificação (HbS), A hemoglobina S, como 90% de outras hemoglobinas anormais, resulta da substituição de um único aminoácido na cadeia globina.
Na anemia falciforme, os glóbulos podem alterar sua consistência e seu formato, tornando-se rígidos. Nestes casos, podem também agrupar-se e formar tampões, dificultando a circulação sanguínea e, consequentemente, a oxigenação dos tecidos. Assim, as manifestações clínicas da doença falciforme são anemia crónica por destruição as hemácias e os fenomenos trombóticos muitas vezes acompanhados de uma dor intensa. Além de provocar palidez, dificuldades em respirar, e batidas do coração aceleradas, infecções agudas, como meningite, inflamação do baço e derrames cerebrais.
Os principais sintomas são:
Crises dolorosas: dor em ossos, músculos e juntas associadas ou não a infecções, exposições ao frio, esforços, etc;
Palidez, cansaço fácil, icterícia (cor amarelada visível principalmente no branco do olho);
Úlceras (feridas) nas pernas;
Maior tendência a infecções.
Doença de Wilson
A Doença de Wilson é uma doença hereditária recessiva, isto significa que uma pessoa precisa receber um gene anormal do pai e da mãe para ser um doente de Wilson: a doença só acontece se a pessoa tiver dois genes anormais.
Cada um dos pais tem que ser um portador do gene anormal. Se ambos os pais forem portadores do gene da Doença de Wilson, então a hipotese de a criança ter a doença é de 25%.
Muitos casos da Doença de Wilson ocorrem devido à mutação espontânea do gene. Muitos são simplesmente transmitidos de geração para geração. Um número significativo de casos não tem uma história familiar da doença.As pessoas com apenas um gene anormal são chamadas de portadoras.
Elas não desenvolvem a doença e nem precisam de tratamento. Mais de trinta mutações foram identificadas. Por isso, tem sido difícil encontrar um único teste genético para diagnosticar a doença. Mas se numa determinada família a mutação for identificada, um diagnóstico genético é possível. Isso pode ajudar no caso de um parente assintomático, de modo que possa ser tratado antes que fique doente ou que seja fisicamente prejudicado. No futuro, um teste genético possibilitará um diagnóstico pré-natal.
Distrofia muscular
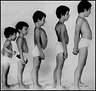
A distrofia muscular progressiva é uma doença de carácter hereditário, tendo como principal característica a degeneração da membrana que envolve a célula muscular, causando a sua morte.
Distrofia muscular é o termo amplo usado para designar um grupo de doenças genéticas que afectam os músculos causando fraqueza. Essa fraqueza muscular, dependendo do tipo de distrofia, afecta grupos de músculos diferentes e tem velocidade de degeneração também diferente.
De uma forma geral a distrofia muscular é considerada rara, sendo a distrofia muscular de Duchenne a forma mais comum dentre destas, afectando cerca de um em cada três mil meninos.
Diagnóstico
De uma forma geral o aparecimento dos primeiros sintomas da distrofia são percebidos pelos próprios pais, o que no caso de Duchenne ou Becker costuma ocorrer na faixa dos três aos cinco anos de idade.
Normalmente nota-se um atraso de desenvolvimento com relação às outras crianças da mesma idade, desequilibram-se e caem com facilidade, tem dificuldades para correr, subir escadas, levantar-se do chão e cansam mais rapidamente. Começando também a caminhar na ponta dos pés, devido ao surgimento de contraturas nos tendões de Aquiles (o que faz com que tenham um gingado incomodo ao andar)
Para concluir-se um diagnóstico são necessários alguns testes específicos. Primeiramente verificam-se os níveis de uma enzima muscular chamada creatina Kinase que tem níveis relativamente baixos no sangue em condições normais, mas aumentam muito sua concentração quando ocorre a degeneração muscular .
Síndrome de Marfan
Descrita inicialmente por Marfan, trata-se de uma doença do tecido conjuntivo caracterizada por McKusick. Os indivíduos afectados têm estaturas altas e com proporções corporais alongadas.
Diagnóstico
Trata-se de uma doença de hereditariedade autossómica dominante, no entanto 15% dos casos são mutações de novo.
Os critérios de diagnóstico de certeza nos indivíduos com história familiar da doença requerem uma manifestação maior para o diagnóstico. Naqueles em que não existem antecedentes familiares, pelos menos duas alterações maiores e o envolvimento de outro sistema devem estar presentes para um diagnóstico correcto.
Tratamento
É raro o diagnóstico no período pré-natal. Alguns doentes só têm sinais sugestivos da doença na pré-adolescência, enquanto outros são adultos assintomáticos mas com um familiar afectado. Perante a suspeita do diagnóstico do síndrome de Marfan e independentemente da idade, deve ser realizado um exame cardiológico e oftalmológico pormenorizado a fim de permitir a identificação e o tratamento adequado das lesões . O tratamento destas complicações tanto pode ser farmacológico como cirúrgico. É importante evitar desportos de alta competição e de contacto, assim como o exercício isométrico. Do ponto de vista da doença cardíaca, mesmo na ausência de alterações evidentes poderá ser necessário iniciar o tratamento farmacológico. As medidas preventivas em doentes com o síndrome de Marfan incluem avaliações periódicas dos olhos, coração e esqueleto.
posted by Teresantunes @ 1:40 PM
1 comments
![]()

